
Abstracto
Antecedentes La enfermedad pulmonar intersticial (EPI) infantil comprende un espectro de EPI raras que afectan a lactantes, niños y adolescentes. Nintedanib es un tratamiento autorizado para la fibrosis pulmonar en adultos. Los objetivos principales del ensayo InPedILD fueron determinar la exposición a la dosis y la seguridad de nintedanib en niños y adolescentes con EPI fibrosante.
Métodos Los pacientes de 6 a 17 años con EPI fibrosante en la tomografía computarizada de alta resolución y enfermedad clínicamente significativa se aleatorizaron 2:1 para recibir nintedanib o placebo durante 24 semanas y luego nintedanib de etiqueta abierta. La dosificación se basó en una escala alométrica dependiente del peso. Los criterios de valoración coprimarios fueron el área bajo la curva de concentración plasmática-tiempo en estado estacionario (AUCτ, ss) en las semanas 2 y 26 y la proporción de pacientes con eventos adversos emergentes del tratamiento en la semana 24.
Resultados 26 pacientes recibieron nintedanib y 13 pacientes recibieron placebo. La media geométrica (coeficiente geométrico de variación) AUCτ, ss para nintedanib fue de 175 µg·h·L−1 (85,1%) en pacientes de 6 a 11 años y 160 µg·h·L−1 (82,7%) en pacientes de 12 a 17 años. En el período doble ciego, se informaron eventos adversos en el 84,6% de los pacientes en cada grupo de tratamiento. Dos pacientes suspendieron nintedanib debido a eventos adversos. Se notificó diarrea en el 38,5 % y el 15,4 % de los grupos de nintedanib y placebo, respectivamente. Media ajustada±se los cambios en el porcentaje previsto de capacidad vital forzada en la semana 24 fueron de 0,3±1,3 % en el grupo de nintedanib y de −0,9±1,8 % en el grupo de placebo.
Conclusiones En niños y adolescentes con EPI fibrosante, un régimen de dosificación basado en el peso dio como resultado una exposición a nintedanib similar a la de los adultos y un perfil de seguridad aceptable. Estos datos proporcionan una base científica para el uso de nintedanib en esta población de pacientes.
Abstracto
Los resultados del ensayo aleatorizado controlado con placebo InPedILD respaldan una evaluación positiva de la relación riesgo-beneficio del uso de nintedanib en niños y adolescentes con EPI fibrosante https://bit.ly/3qnAhUM
Introducción
La enfermedad pulmonar intersticial infantil (chILD, por sus siglas en inglés) comprende un espectro de trastornos pulmonares raros y heterogéneos que afectan a lactantes, niños y adolescentes que pueden estar asociados con una morbilidad significativa [1, 2]. La fisiopatología de chILD a menudo implica un componente genético, a veces combinado con lesiones relacionadas con la exposición o desregulación autoinmune, aunque en algunos casos la etiología sigue siendo desconocida. [1, 3–6]. Algunos niños con ILD desarrollan fibrosis pulmonar. Al igual que en los adultos, en la EPI infantil, la fibrosis pulmonar implica daño tisular, depósito excesivo de matriz extracelular y remodelación aberrante del pulmón, aunque histológicamente no se observan focos fibroblásticos en niños con fibrosis. [1, 7]. En ausencia de medicamentos autorizados para el tratamiento de chILD, el tratamiento generalmente implica inmunomodulación [8]pero falta evidencia para apoyar este enfoque.
Nintedanib es un inhibidor intracelular de tirosina quinasas que inhibe procesos fundamentales para la progresión de la fibrosis pulmonar [9, 10]. Los ensayos controlados aleatorios demostraron que en adultos con fibrosis pulmonar idiopática, otras formas de fibrosis pulmonar progresiva y EPI fibrosante asociada con la esclerosis sistémica, nintedanib redujo consistentemente la tasa de disminución de la capacidad vital forzada (FVC), una medida sustituta de la progresión de la EPI en adultos. [11]en comparación con el placebo, con un perfil de eventos adversos caracterizado predominantemente por eventos gastrointestinales [12–15]. Sobre la base de la eficacia clínica de nintedanib en adultos y las supuestas similitudes en la fisiopatología de la remodelación pulmonar fibrótica en adultos y niños, se postuló que nintedanib puede proporcionar un beneficio similar en una población pediátrica con fibrosis pulmonar clínicamente significativa o progresiva. Como no se consideró factible un ensayo confirmatorio de la eficacia de nintedanib en pacientes pediátricos, el ensayo InPedILD (ClinicalTrials.gov: NCT04093024; Registro de ensayos clínicos de la UE: EudraCT 2018-004530-14) se diseñó con los objetivos principales de evaluar la dosis-exposición y la seguridad de nintedanib en niños y adolescentes con EPI fibrosante. Además, se recopilaron datos sobre los criterios de valoración de la eficacia para respaldar la evaluación de la relación riesgo-beneficio de nintedanib en la población pediátrica.
Métodos
Pacientes
El ensayo InPedILD inscribió a niños o adolescentes de 6 a 17 años con EPI fibrosante en una tomografía computarizada de alta resolución (TCAR) realizada ≤12 meses antes de la selección, confirmada por revisión central (material suplementario), CVF ≥25 % del valor teórico y enfermedad clínicamente significativa [16]. La enfermedad clínicamente significativa se definió como una puntuación de Fan ≥3 [17] o evidencia documentada de progresión clínica en cualquier período de tiempo. La puntuación de Fan evalúa la gravedad de la enfermedad en niños y adolescentes con EPI en función de los síntomas, la saturación de oxígeno y la hipertensión pulmonar; las puntuaciones van del 1 al 5, y las puntuaciones más altas indican una mayor gravedad de la enfermedad [17]. La evidencia de progresión clínica se basó en una disminución relativa de la CVF ≥10 % del valor teórico, una disminución relativa de la CVF del 5 al 10 % del valor teórico con empeoramiento de los síntomas, empeoramiento de la fibrosis en la TACAR u otras medidas de empeoramiento clínico atribuidas a la fibrosis pulmonar progresiva (p.ej aumento del requerimiento de oxígeno o disminución de la capacidad de difusión). Los criterios clave de exclusión se enumeran en el material suplementario.
Diseño de prueba
El ensayo InPedILD fue un ensayo de fase 3 realizado en 43 sitios en 21 países [16]. Después de un período de selección de 4 semanas, los pacientes fueron aleatorizados 2:1 para recibir nintedanib o placebo utilizando tecnología de respuesta interactiva, estratificados por grupo de edad (6 a 11 y 12 a 17 años) (Figura 1). La dosificación se basó en una escala alométrica dependiente del peso. Las dosis iniciales fueron 50, 75, 100 o 150 mg dos veces al día y la dosis se ajustó durante el tratamiento en función del peso del paciente (tabla 1). Se permitieron reducciones de dosis a la dosis siguiente e interrupciones del tratamiento para controlar los eventos adversos. La dosis más baja fue de 25 mg dos veces al día. Se permitió volver a escalar la dosis dentro de las 4 semanas posteriores a la reducción de la dosis para los eventos adversos que se consideraron relacionados con el fármaco del ensayo o dentro de las 8 semanas para los eventos adversos que no se consideraron relacionados con el fármaco del ensayo.
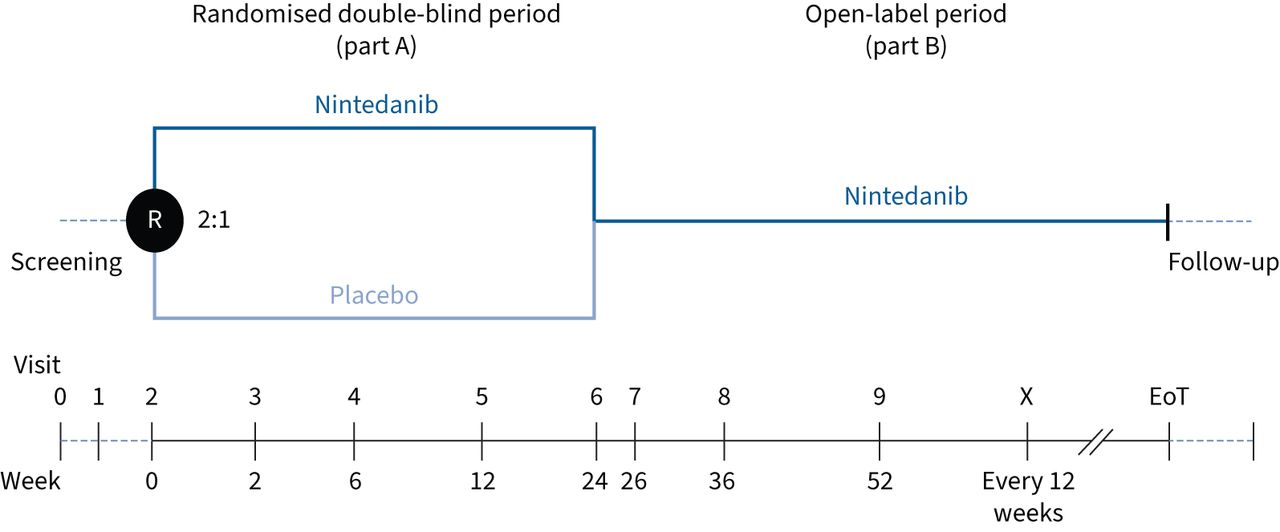
Diseño de prueba. R: aleatorización; EoT: fin del tratamiento.
Dosificación y ajustes de dosis
El ensayo consistió en un período doble ciego controlado con placebo de 24 semanas seguido de un período variable durante el cual todos los pacientes recibieron nintedanib de etiqueta abierta (Figura 1). A los pacientes que interrumpieron prematuramente el fármaco del estudio se les pidió que asistieran a todas las visitas según lo planeado originalmente. Una vez que ≥30 pacientes (incluidos ≥20 pacientes de 12 a 17 años) completaron el muestreo farmacocinético en la semana 26 o interrumpieron prematuramente el ensayo, se cerró el reclutamiento. Luego de la confirmación de que se habían recopilado suficientes datos farmacocinéticos, se programó a todos los pacientes para una visita de finalización del tratamiento, luego de lo cual ingresaron a un período de seguimiento de 4 semanas o pasaron a un ensayo de extensión abierto en el que todos los pacientes recibieron nintedanib (InPedILD-ON: ClinicalTrials.gov: NCT05285982; Registro de ensayos clínicos de la UE: EudraCT 2020-005554-23).
Un comité directivo brindó asesoramiento científico sobre el diseño del ensayo. Un comité de control de seguridad revisó los datos farmacocinéticos y de seguridad para determinar el perfil de seguridad y el riesgo-beneficio de nintedanib, y asesorar sobre la modificación de la dosis, evaluaciones adicionales y la idoneidad de más inscripciones y la continuación del ensayo. Se estableció un comité de adjudicación para adjudicar las muertes por causas cardíacas, respiratorias u otras y revisar los eventos adversos categorizados como eventos cardiovasculares adversos mayores. El ensayo se llevó a cabo de conformidad con el protocolo, los principios de la Declaración de Helsinki y la Guía Tripartita Armonizada de Buenas Prácticas Clínicas de la Conferencia Internacional sobre Armonización, la directiva de la UE 2001/20/EC/Reglamento de la UE 536/2014 y otras directrices pertinentes. El ensayo se inició en cada sitio (listado en el material suplementario) luego de la aprobación por parte de la respectiva junta de revisión institucional/comité de ética independiente y autoridad competente de acuerdo con las regulaciones nacionales e internacionales. El consentimiento informado por escrito y el asentimiento (cuando corresponda) se obtuvieron antes del ingreso al ensayo.
Puntos finales
Los criterios de valoración coprimarios fueron el área bajo la curva de concentración plasmática-tiempo en estado estacionario (AUCτ, ss) en la semana 2 de tratamiento con nintedanib (es decir en la semana 2 en pacientes aleatorizados para recibir nintedanib y en la semana 26 en pacientes aleatorizados para recibir placebo durante 24 semanas seguido de nintedanib) y la proporción de pacientes con eventos adversos emergentes del tratamiento en la semana 24, que se definieron como eventos con inicio a partir del primera toma del fármaco del ensayo hasta el día anterior a la primera toma de nintedanib de etiqueta abierta o la última toma del tratamiento aleatorizado más 28 días (lo que ocurra primero). Los eventos adversos se codificaron de acuerdo con el Diccionario médico para actividades regulatorias (MedDRA) versión 25 (www.meddra.org).
Los hallazgos patológicos en las imágenes óseas y el retraso en el crecimiento de la raíz dental en las imágenes dentales se definieron como eventos adversos de especial interés. Los criterios de valoración de seguridad secundarios incluyeron la proporción de pacientes con hallazgos patológicos de cartílagos de crecimiento epifisario en imágenes y hallazgos patológicos en el examen dental o en imágenes en la semana 24. Las imágenes de seguimiento de cartílagos de crecimiento epifisario solo se realizaron en pacientes con fisis abiertas. El cambio desde el inicio en la puntuación z de talla para la edad en la semana 24 fue otro criterio de valoración.
Los criterios de valoración secundarios de la eficacia incluyeron cambios en el % pred de FVC, saturación de oxígeno periférico (Scorreos2) en reposo, la distancia de la prueba de caminata de 6 minutos (6MWT) y las puntuaciones del cuestionario de calidad de vida pediátrica (PedsQL) en la semana 24. La espirometría se realizó utilizando espirómetros estandarizados suministrados a cada sitio y de acuerdo con las pautas de la American Thoracic Society/European Respiratory Society [18]. Los valores de FVC % pred se calcularon usando las ecuaciones publicadas por la Iniciativa Global de Función Pulmonar [19]. El cuestionario PedsQL comprendía el informe de niños pequeños (5 a 7 años), el informe de niños (8 a 12 años) y el informe de adolescentes (>12 años), así como un informe de padres para cada rango de edad; cada uno de estos puntajes varía de 0 a 100, y los puntajes más altos indican una mejor calidad de vida relacionada con la salud [20].
Análisis
Para el análisis farmacocinético primario, AUCτ, ss se calculó utilizando análisis no compartimentales y compartimentales y estadística descriptiva. En pacientes aleatorizados a nintedanib, el AUCτ, ss en la semana 2 se utilizó; si faltaba este valor, se utilizó el valor de la semana 26. En pacientes aleatorizados a placebo, el AUCτ, ss en la semana 26 (correspondiente a la semana 2 de tratamiento con nintedanib). Los análisis farmacocinéticos se realizaron en pacientes que recibieron ≥1 dosis del medicamento de prueba y proporcionaron datos evaluables para ≥1 puntos finales farmacocinéticos sin violaciones del protocolo relevantes para la evaluación de la farmacocinética. Los análisis de seguridad se realizaron en pacientes que recibieron ≥1 dosis del medicamento de prueba. Cambios medios ajustados desde el inicio en FVC % pred, Scorreos2, la distancia 6MWT, las puntuaciones del cuestionario PedsQL y la puntuación z de la talla para la edad en la semana 24 se analizaron mediante un modelo mixto para medidas repetidas, con efectos categóricos fijos del tratamiento en cada visita y grupo de edad y efectos continuos fijos de referencia en cada visita y efecto aleatorio para el paciente. Los eventos adversos se presentan de forma descriptiva.
El tamaño de la muestra se calculó en base a la evaluación del punto final farmacocinético primario y la necesidad de estimar el parámetro de aclaramiento con la precisión adecuada. Suponiendo un coeficiente de variación (CV) del 70,8 % (basado en el CV geométrico (gCV) del aclaramiento aparente de nintedanib en el plasma en estado estacionario (CL/Fss) después de extravascular…


{kind=link}