Resumen
Antecedentes
La linfangiomatosis pulmonar (PL) es una enfermedad ultrarara caracterizada por una infiltración difusa del pulmón, la pleura y/o el mediastino por una proliferación linfática anormal. No se establecen enfoques de diagnóstico o tratamiento consentidos. Por lo tanto, nuestro objetivo fue recopilar datos sobre diagnósticos y tratamientos en una cohorte de pacientes con PL de un centro terciario para enfermedades pulmonares raras.
Métodos
Los datos clínicos, radiológicos y de resultados de pacientes con PL se recopilaron retrospectivamente.
Resultados
12 pacientes fueron diagnosticados entre 1996 y 2022 en nuestro centro. La PL se diagnosticó con mayor frecuencia en mujeres (58%), nunca fumadoras (75%) y pacientes más jóvenes (edad media 42 años). Los principales síntomas clínicos fueron hemo y quiloptisis (58%) y disnea de esfuerzo (83%). La función pulmonar fue mayoritariamente restrictiva (media VC 59%) con DLCO alterada (media 65%). La evaluación radiológica mostró principalmente afectación mediastínica (83%), derrame pleural (67%), engrosamiento pleural (67%) y engrosamiento de la pared bronquial (67%), mientras que los cambios intersticiales fueron raros. El diagnóstico se confirmó mediante criobiopsia quirúrgica o transbronquial. Ocho pacientes fueron tratados con sirolimus, 3 de ellos combinados con una intervención quirúrgica y en un caso la intervención quirúrgica fue necesaria 9 meses después del inicio de sirolimus. Se demostró mejoría clínica y radiológica en todos los pacientes tratados con sirolimus. 1 paciente recibió un trasplante de pulmón por progresión de la enfermedad. Las tasas de supervivencia fueron del 90% después de un seguimiento medio de al menos 3 meses.
Conclusión
Esta serie de casos ilustra la variabilidad de la presentación clínica de PL. Entre nuestros pacientes, los tratados con sirolimus mostraron una importante mejoría clínica, funcional y radiológica. Sin embargo, se necesita más investigación para comprender la patogénesis de la linfangiomatosis y poder establecer enfoques terapéuticos.
Fondo
La linfangiomatosis pulmonar es una enfermedad ultrarara que suele presentarse en la infancia y la edad adulta. El término ultrararo fue introducido inicialmente por el Instituto Nacional para la Excelencia en Salud y Atención Médica para medicamentos con indicación para enfermedades que tienen una prevalencia <1 por 50.000 personas (1), sin embargo no está definido legalmente.
Se postula que la PL es congénita y afecta a ambos sexos por igual. Los síntomas son variables, desde pacientes asintomáticos hasta pacientes con dificultad respiratoria grave que provocan la muerte (2). La mayoría de los pacientes presentan disnea de esfuerzo, hemoptisis, derrame quiloso o dolor torácico (3). Las pruebas de función pulmonar pueden mostrar trastornos de la ventilación tanto obstructivos como restrictivos, además de insuficiencia respiratoria (4). La tomografía computarizada (TC) de los pulmones puede mostrar engrosamiento pleural, derrame pleural, engrosamiento septal y peribroncovascular, así como infiltración de tejidos blandos mediastínicos (5). La biopsia generalmente se obtiene mediante toracoscopia videoasistida o biopsias transbronquiales (6). Sin embargo, la mayoría de los casos dentro de la literatura fueron diagnosticados mediante resección toracoscópica en cuña. Histológicamente, los linfangiomas con células endoteliales linfoides son positivos para CD-31 y D2-40 (7). El factor de crecimiento endotelial vascular (VEGF)-D es un factor linfangiogénico establecido (8) probablemente desempeñando un papel importante en la patogénesis de la linfangiomatosis (9). Actualmente no existe un régimen de tratamiento establecido ni una terapia causal. Los tratamientos actuales tienen como objetivo reducir el aumento de la secreción linfática. Los informes de casos mostraron cierta eficacia para varios fármacos, incluido el inhibidor de mTOR sirolimus, el betabloqueante no selectivo propranolol, tratamientos quimioterapéuticos y quirúrgicos y radioterapia (2, 9).
Métodos
Analizamos nuestra base de datos de pacientes con enfermedades pulmonares raras diagnosticadas entre 1996 y 2022 para linfangiomatosis pulmonar confirmada. Las TC iniciales de los casos identificados fueron obtenidas y revisadas nuevamente por un radiólogo torácico experimentado. Variables demográficas (edad; sexo; disnea; tos; dolor torácico; tabaquismo), pruebas de función pulmonar, capacidad de difusión pulmonar de monóxido de carbono (DLCO); Se recopilaron patrones histopatológicos y formas de tratamientos, así como resultados.
También realizamos una investigación bibliográfica en PubMed Central® (PMC) para obtener un número estimado de informes de casos sobre linfangiomatosis pulmonar y afectación pulmonar en linfangiomatosis, respectivamente, durante los últimos 40 años. El término de búsqueda utilizado fue “linfangiomatosis pulmonar”. Todos los resultados fueron revisados críticamente para identificar informes relevantes.
Resultados
Durante 1996 a 2022, doce pacientes fueron diagnosticados con linfangiomatosis pulmonar en nuestro centro terciario de EPI. Dos de ellos fueron diagnosticados con probable linfangiomatosis pulmonar debido a una histología no concluyente. La linfangiomatosis pulmonar se diagnosticó con mayor frecuencia en mujeres (58%), nunca fumadoras (75%) y pacientes más jóvenes (edad media 42 años). Los principales síntomas clínicos fueron hemo y quiloptisis (58%) y disnea de esfuerzo (83%). Dos pacientes sufrieron hemoptisis y quiloptisis, y un paciente sufrió insuficiencia ventilatoria. La función pulmonar fue restrictiva (media VC 59%) con DLCO alterada (media 65%) en la mayoría de los casos (Tabla 1).
Tabla 1 Características de los pacientes al primer diagnósticomesa de tamaño completoLa evaluación radiológica mostró afectación mediastínica en todos los pacientes (Figs. 1a y b, 2, 3) excepto uno. Derrame pleural, engrosamiento pleural, engrosamiento de la pared bronquial y engrosamiento septal (Figs. 2, 3) fueron hallazgos radiológicos comunes (cada 67%). Todos los hallazgos radiológicos y su frecuencia se muestran en la Fig. 3.
Figura 11a y 1b: Ecografía endobronquial de masa mediastínica que muestra vasos no sanguíneos dilatados y de bajo flujo
Imagen a tamaño completoFigura 2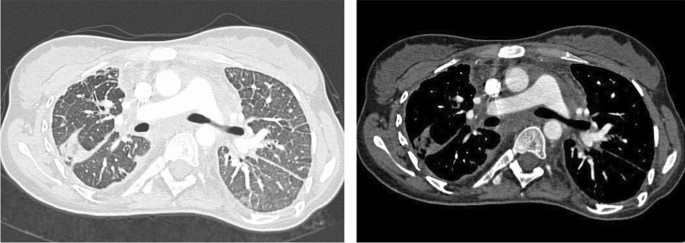
TC-Tórax: masa mediastínica, engrosamiento pleural y engrosamiento septal en PL
Imagen a tamaño completofigura 3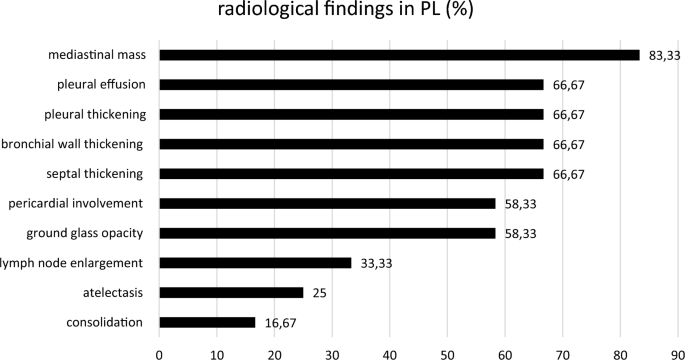
Hallazgos radiológicos en PL y su incidencia.
Imagen a tamaño completoEl diagnóstico se confirmó mediante biopsia quirúrgica o biopsia transbronquial en todos los pacientes. La histología mostró ectasia vascular pleural y peribronquial con linfangioma, D2-40 positivo, en la mayoría de los casos.
Nueve pacientes fueron tratados con sirolimus, 4 de ellos combinados con intervención quirúrgica/resección. Se demostró mejoría clínica y/o radiológica en todos los pacientes tratados con sirolimus durante al menos 3 meses de seguimiento (Tabla 2; Higo. 4).
Tabla 2 tratamientos y resultadomesa de tamaño completoFigura 4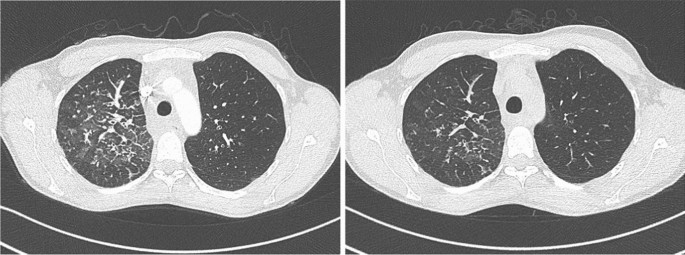
TC-Tórax antes y después de 3 meses de tratamiento con sirolimus
Imagen a tamaño completoUn paciente mostró una mejoría del derrame pericárdico quiloso asociado a PL debido al tratamiento con sirolimus (Fig. 5).
figura 5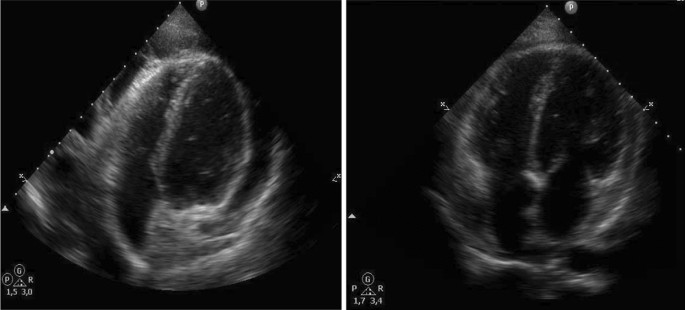
Ecocardiografía en un paciente con PL y derrame pericárdico quiloso asociado antes y después de 8 meses de tratamiento con sirolimus
Imagen a tamaño completoUno de los pacientes que recibió sirolimus en combinación con propranolol presentó deterioro a los 9 meses de inicio con derrame pleural progresivo y síntomas de insuficiencia cardíaca. Luego se realizó intervención quirúrgica (adhesiolisis, pleurodesis con talco, pericardiectomía) seguida de tratamiento farmacológico solo con sirolimus. 3 meses después se logró una mejoría clínica, de la función pulmonar y radiológica. En 1 paciente se planificó el inicio de sirolimus pero no se inició por esperar a la aprobación de un uso off-label y luego pérdida del seguimiento. 1 paciente recibió un trasplante de pulmón debido a la progresión de la enfermedad después del tratamiento quirúrgico inicial, pero murió poco después debido a una complicación. No se pudo evaluar el resultado de dos pacientes debido a pérdidas en el seguimiento. Las tasas de supervivencia fueron del 92% después de un seguimiento medio de al menos 3 meses (Tabla 2).
El número estimado de informes de casos sobre linfangiomatosis pulmonar y afectación pulmonar de linfangiomatosis respectivamente en Pubmed Central® (PMC) fue de 71 en los últimos 40 años (Suplemento).
Discusión
La linfangiomatosis pulmonar es causada por la proliferación de vasos linfáticos en los tejidos blandos (2). Nuestra serie de casos ilustra la variabilidad de la presentación clínica y las afecciones de diferentes sitios del tórax en la linfangiomatosis pulmonar. Debido a la rareza de la linfangiomatosis, es difícil establecer una estrategia de tratamiento basada en evidencia. La mayoría de los tratamientos son de apoyo y tienen como objetivo descomprimir las estructuras adyacentes y la acumulación de líquido quiloso. Aquí ofrecemos más información sobre esta enfermedad ultrarara añadiendo más conocimientos sobre el diagnóstico y las posibilidades terapéuticas, especialmente en el tratamiento con sirolimus. Entre nuestros pacientes, los tratados con sirolimus mostraron una importante mejoría morfológica clínica, pulmonar y TC con un nivel terapéutico de 5 ng/ml. Esto está en línea con una revisión sistemática reciente que informa que el tratamiento con el inhibidor de mTOR sirolimus fue un tratamiento eficaz y seguro para pacientes con anomalías vasculares complicadas, incluida la linfangiomatosis, refractaria a otras terapias (10). Como efecto subyacente del sirolimus, se discute que sirolimus se une al receptor 3 de VEGF en la superficie del endotelio linfático (11). Nuestros datos también están en línea con algunos datos limitados de informes de casos que confirman un tratamiento exitoso con sirolimus en varios casos (12,13,14,15). Los informes sobre otras terapias en PL son escasos. Una posible opción de tratamiento podría ser la radioterapia mediante la fibrosis del endotelio linfático inducida por la radiación, que conduce a la destrucción de los vasos linfáticos y provoca una regresión de las lesiones durante varios meses.16). Esta opción terapéutica también fue elegida en uno de los pacientes, donde la combinación de radioterapia y sirolimus finalmente condujo a una mejora clínica significativa. Con respecto al tratamiento quirúrgico, nuestros datos sugieren que la resección puede tener un efecto en lesiones pulmonares o mediastínicas localizadas. Otros tratamientos quirúrgicos son la pleurodesis, la pleurectomía parietal y la ligadura del conducto torácico (17). Sin embargo, las manifestaciones de la enfermedad pueden recaer después de los procedimientos quirúrgicos, ya que el tejido enfermo restante puede provocar…