Resumen
Antecedentes
La hipertensión arterial pulmonar (HAP) es una enfermedad cardiopulmonar crónica potencialmente mortal. Sin embargo, hay una escasez de estudios que reflejen los biomarcadores disponibles a partir de perfiles de expresión génica separados en la HAP.
Métodos
Los conjuntos de datos GSE131793 y GSE113439 se combinaron para análisis posteriores y se eliminaron los efectos de lote. Luego se realizó un análisis bioinformático para identificar genes expresados diferencialmente (DEG). Luego se utilizó un análisis de red de coexpresión génica ponderada (WGCNA) y un análisis de red de interacción proteína-proteína (PPI) para filtrar aún más los genes centrales. El análisis de enriquecimiento funcional de los genes de intersección se realizó utilizando Gene Ontology (GO), Disease Ontology (DO), Kyoto encyclopedia of genes and genomes (KEGG) y análisis de enriquecimiento de conjunto de genes (GSEA). El nivel de expresión y el valor diagnóstico de la expresión del gen central en pacientes con hipertensión arterial pulmonar (HAP) también se analizaron en los conjuntos de datos de validación GSE53408 y GSE22356. Además, la expresión del gen objetivo se validó en los pulmones de un modelo de rata con hipertensión pulmonar (HP) inducida por monocrotalina (MCT) y en el suero de pacientes con HAP.
Resultados
Se identificaron un total de 914 genes expresados diferencialmente (GED), con 722 genes sobreexpresados y 192 sobreexpresados. El módulo clave relevante para la HAP se seleccionó utilizando WGCNA. Al combinar los GED y el módulo clave de WGCNA, se seleccionaron 807 genes. Además, el análisis de la red de interacción proteína-proteína (PPI) identificó a HSP90AA1, CD8A, HIF1A, CXCL8, EPRS1, POLR2B, TFRC y PTGS2 como genes centrales. Los conjuntos de datos GSE53408 y GSE22356 se utilizaron para evaluar la expresión de TFRC, que también mostró un sólido valor diagnóstico. Según el análisis de enriquecimiento de GSEA, las funciones y vías biológicas relevantes para la HAP se enriquecieron en pacientes con altos niveles de TFRC. Además, se encontró que la expresión de TFRC estaba regulada positivamente en los tejidos pulmonares de nuestro modelo experimental de rata con HAP en comparación con los de los controles, y se llegó a la misma conclusión en el suero de los pacientes con HAP.
Conclusiones
Según nuestro análisis bioinformático, el aumento observado de TFRC en el tejido pulmonar de pacientes humanos con HAP, tal como lo indican los datos transcriptómicos, es consistente con las alteraciones observadas en pacientes con HAP y modelos de roedores. Estos datos sugieren que TFRC puede servir como un posible biomarcador para la HAP.
Introducción
La hipertensión arterial pulmonar (HAP) es un trastorno complejo y progresivo que se caracteriza por una presión arterial pulmonar elevada que conduce a insuficiencia ventricular derecha (1). Aunque se han desarrollado numerosas terapias dirigidas para la HAP con el fin de aliviar eficazmente los síntomas, esta enfermedad grave sigue estando asociada a un mal pronóstico. La tasa de supervivencia a cinco años para los pacientes recién diagnosticados es de tan solo el 61,2 % (2). El diagnóstico temprano de la HAP es importante para los pacientes, por lo tanto, existe una necesidad urgente de identificar el mecanismo potencial de la HAP e identificar biomarcadores relacionados.
Los recientes avances en bioinformática han permitido una comprensión más matizada de la base molecular de los HAP (3,4,5). Los métodos de análisis bioinformático pueden ayudar a identificar genes candidatos relacionados con el desarrollo de la HAP, ofreciendo una valiosa guía para biomarcadores de diagnóstico o pronóstico y objetivos terapéuticos (6, 7). Sin embargo, muchos estudios actuales sólo hacen predicciones teóricas sin validar experimentalmente su fiabilidad (5, 8).
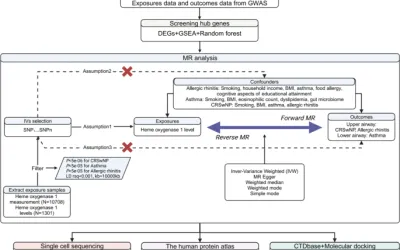
Primero fusionamos los datos de microarrays de PAH de las bases de datos públicas Gene Expression Omnibus (GEO) GSE131793 y GSE113439, para eliminar las diferencias entre lotes. Después de evaluar la calidad de los datos sin procesar, se identificaron los genes expresados diferencialmente (DEG) utilizando el paquete limma en la versión 4.3.2 del software R.http://www.r-project.org/). El desarrollo de redes de coexpresión ha facilitado la creación de métodos de detección genética basados en redes, que pueden utilizarse para identificar posibles biomarcadores y objetivos terapéuticos (9). Se empleó WGCNA para identificar genes asociados con el fenotipo clínico. Los DEG mencionados anteriormente, que se superponen con los módulos clave de WGCNA, se utilizaron con el fin de realizar un análisis de enriquecimiento funcional y de vías mediante el uso de GO, DO, KEGG y GSEA. Posteriormente, se construyó una red de interacción proteína-proteína (PPI) utilizando estos genes. Posteriormente, se identificaron los genes centrales mediante el uso del software Cytoscape. La red PPI se intersectó con los 20 genes centrales principales del análisis de red biológica con cuatro algoritmos, lo que resultó en la selección de 8 genes: HSP90AA1, CD8A, HIF1A, CXCL8, EPRS1, POLR2B, TFRC y PTGS2. Después de la verificación de dos conjuntos de datos independientes y la validación experimental, se identificó a TFRC como un biomarcador molecular potencial en HAP. La siguiente sección describe los materiales y métodos utilizados en este estudio.
Materiales y métodos
Análisis de datos de microarrays e identificación de genes expresados diferencialmente
Los conjuntos de datos de expresión genética GSE131793 (10), GSE113439 (11), GSE53408 (12) y GSE22356 (13) fueron seleccionados para este estudio. Todos los datos de microarrays de expresión génica se obtuvieron de la base de datos GEO en forma de una matriz de expresión génica estandarizada y de calidad controlada (https://www.ncbi.nlm.nih.gov/geo/). El conjunto de datos GSE131793 incluyó 10 muestras de pacientes con HAP y 10 muestras de controles normales. El conjunto de datos GSE113439 comprende 15 muestras de HAP y 11 muestras de control. Los conjuntos de datos GSE53408 y GSE22356 se seleccionaron como conjuntos de validación, que contienen 12 muestras de HAP y 11 muestras de control y 18 muestras de HAP y 20 muestras de control, respectivamente. Los conjuntos de datos de microarray GSE131793 y GSE113439 se integraron como conjuntos de datos de entrenamiento después de excluir la variación entre lotes utilizando el paquete de análisis de variables sustitutas (SVA) (14). Se empleó un análisis de componentes principales (PCA) bidimensional para ilustrar la disparidad entre los grupos de lotes pre y post-SVA. La normalización de los datos y la corrección de fondo se realizaron utilizando el método de promedio de multiarray robusto (RMA). Las sondas de microarray se anotaron utilizando archivos de anotación, lo que resultó en la eliminación de cualquier sonda genética que no estuviera alineada con un gen específico. En el caso de que varias sondas representaran el mismo símbolo genético, se utilizó el valor promedio como una medida representativa. La detección de genes para la expresión diferencial se realizó utilizando el paquete «limma», aplicando un umbral de significancia de P < 0,05 y un |log2 fold change (FC)|> 0,5. El paquete ggplot2 (15) se utilizó para generar un gráfico de volcanes de los DEG, y se utilizó el paquete pheatmap para construir un mapa de calor de los DEG.
Módulos identificados como relacionados con HAP a través del análisis de WGCNA
Se utilizó la herramienta WGCNA en R para construir una red de coexpresión ponderada de los conjuntos de datos fusionados (9). Para construir la red libre de escala, se utilizó la función pickSoftThreshold para seleccionar potencias blandas b = 10. Se generó una matriz de adyacencia, que luego se transformó en una matriz de superposición topológica (TOM) y la matriz de disimilitud correspondiente (1-TOM). Se construyó un diagrama de árbol de agrupamiento jerárquico de la matriz 1-TOM para clasificar patrones de expresión génica similares en diferentes módulos de coexpresión génica. Para identificar módulos funcionales adicionales en la red de coexpresión, se calcularon asociaciones módulo-rasgo entre módulos e información de características clínicas en función de estudios realizados previamente con la intención de identificar aquellos módulos que demostraron coeficientes de correlación altos con características clínicas. Para identificar genes clave expresados diferencialmente (DEG), la herramienta en línea (https://bioinfogp.cnb.csic.es/tools/venny/) se utilizó para construir diagramas de Venn que demuestran la intersección de los DEG y los módulos clave.
Metodología para el análisis de enriquecimiento
Para el análisis de enriquecimiento se utilizaron la ontología genética (GO), la ontología de enfermedades (DO), el análisis de enriquecimiento de conjuntos de genes (GSEA) y la Enciclopedia de Kyoto de genes y genomas (KEGG). El análisis utilizó el 'clusterProfiler' (16) y 'DOSIS' (17) paquetes para realizar análisis de enriquecimiento de GO, KEGG y DO, con un umbral de significancia de P < 0,05.
Construcción y análisis de redes de interacción proteína-proteína (PPI)
Para investigar las interacciones proteína-proteína entre los genes expresados diferencialmente (DEG) identificados en nuestro estudio, utilizamos la base de datos STRING (https://string-db.org/). La red se procesó utilizando el software Cytoscape versión 3.8.2 (https://www.cytoscape.org) para visualización y análisis. Los 20 genes principales identificados mediante análisis de redes biológicas con cuatro algoritmos a través del complemento Cytoscape de CytoHubba.
Establecimiento del modelo de ratón con PH inducido por MCT
Este estudio se adhirió a la Guía para el cuidado y uso de animales de laboratorio (revisada en 1996) de los Institutos Nacionales de Salud de los Estados Unidos (publicación n.º 85-23), y recibió la aprobación del Comité Institucional de Cuidado y Uso de Animales de la Universidad de Jilin, Changchun, China. El estudio empleó ratas Sprague-Dawley, con un peso de entre 180 y 220 g y seis semanas de edad. Las ratas se obtuvieron de Vital River Laboratories Co., Ltd., Beijing, China. Un total de 16 machos se alojaron en un entorno específico libre de patógenos con un ciclo de luz/oscuridad de 12 horas, una temperatura de 25 ± 2 °C y una humedad del 50% ± 5%. Se permitió que los animales se aclimataran durante dos semanas antes de comenzar el experimento. Las ratas fueron asignadas aleatoriamente a dos grupos: un grupo modelo de HAP que recibió una única inyección subcutánea de 60 mg/kg de MCT (Sigma, St. Louis, MO, EE. UU., n = 8), y un grupo de control al que se le administró solución salina (n = 8). Después de un intervalo de tres semanas, todas las ratas fueron pesadas y anestesiadas con una dosis de 60 ml/kg de hidrato de cloral para prepararlas para las evaluaciones posteriores del desarrollo de la hipertensión arterial pulmonar.
Medición hemodinámica, recolección de tejido y análisis histopatológico.
Para evaluar el ventrículo derecho…